
 Galego
GalegoAquellas que afectan al sistema nervioso y a los músculos. Las enfermedades más frecuentes y conocidas por la población son:
La demencia no es un enfermedad específica sino que se trata de un término descriptivo para un conjunto de síntomas que a su vez puede ser causado por diferentes enfermedades que afectan al cerebro. Las personas con demencia tienen alteradas sus capacidades intelectuales de forma significativa, de manera que esto les va a interferir en su actividad normal y relaciones sociales. Igualmente pierden capacidad para resolver problemas y mantener el control sobre sus emociones, con frecuencia pueden experimentar cambios de personalidad y tener problemas de comportamiento, como agitación, delirios y alucinaciones.
Aunque la pérdida de memoria es un síntoma muy común de demencia, perder memoria en sí no significa que una persona tenga demencia. Incluso algunos enfermos no tienen afectada de forma significativa su memoria al inicio del proceso. Los neurólogos sólo diagnostican demencia si dos o más funciones cerebrales cognitivas (memoria, lenguaje, juicio, orientación) están afectadas de forma relevante en una persona con plena consciencia.
Las formas o causas de demencia son múltiples. Aunque la gente identifica demencia con la enfermedad de Alzheimer ésta es únicamente la forma más común, suponiendo un 60-70% de los casos. Otras formas también frecuentes son la demencia vascular (por ejemplo: tras haber sufrido un ictus), la demencia por cuerpos de Lewy y un complejo grupo de enfermedades que originan la demencia frontotemporal (degeneración del lóbulo frontal del cerebro con alteraciones precoces en la personalidad y comportamiento del individuo). En ocasiones los límites entre estas diferentes formas de demencia son difusos, sobre todo al inicio o cuando coexisten varias a la vez, lo que denominamos demencia mixta.
Los neurólogos además hemos de identificar condiciones que pueden causar demencias reversibles, tratables o incluso pseudodemencias, como son los efectos adversos de medicaciones, alteraciones metabólicas y endocrinas, déficits nutricionales, infecciones, intoxicaciones, tumores cerebrales, anoxia e hipoxia cerebral (falta de oxígeno al cerebro), enfermedades del corazón y de los pulmones .
A pesar de ser muy común en personas mayores, nunca debemos pensar que forma parte del envejecimiento normal, y aunque a veces cuesta diferenciar los fallos propios de la edad del inicio de una demencia, el neurólogo tiene herramientas para ello.
La demencia es una de las principales causas de discapacidad y dependencia entre las personas mayores de todo el mundo suponiendo en muchas ocasiones un tremendo sufrimiento para el paciente pero también para sus cuidadores y familiares
EPIDEMIOLOGÍA:LAS CIFRAS
En España hay alrededor de 600.000 personas con demencia y cerca de 400.000 con enfermedad de Alzheimer. La prevalencia es cuatro veces mayor en grupos de mayor edad, en mujeres y en regiones del centro y noroeste de España. Se calcula que en Galicia hay unos 63.000 afectados, pero la falta de registros hace pensar que esta cifra puede ser mayor al haber casos sin diagnosticar.
Galicia es una comunidad envejecida y se espera un crecimiento exponencial en la carga de trastornos neurodegenerativos crónicos como la demencia.
¿CÓMO SE DIAGNOSTICAN?
En la mayor parte de las demencias no hay un test o una prueba que las diagnostique con seguridad. El diagnóstico se debe hacer por médicos expertos y se asienta en tres pilares fundamentales, que son por orden de importancia:
- Los síntomas que refiere el paciente y su familia
- La exploración del enfermo sobre todo la neuropsicológica (test con preguntas que exploran las diferentes capacidades cognitivas)
- Se realizarán estudios siempre encaminados a descartar otras enfermedades pero también existe ya la posibilidad de hacer pruebas que indirectamente nos orientan a la causa de la demencia que padece el enfermo
Un diagnóstico precoz y correcto:
- Beneficia al cuidador: reducen el síndrome del cuidador quemado, la depresión y otros índices de salud mental, mejora la interpretación y el manejo de los trastornos de conducta.
- Y beneficia al enfermo: permite la instauración precoz de un tratamiento especifico, reduce la necesidad de medicación para los trastornos de conducta y mejora las actividades de la vida cotidiana.
TRATAMIENTO
- Farmacólogico: a día de hoy se dispone de medicamentos específicos para el Alzheimer y alguna otra demencia progresiva. Aunque estos fármacos no frenan la enfermedad ni revierten el daño cerebral pueden mejorar ciertos síntomas y lentificar la progresión. Ésto a su vez mejora la calidad de vida del individuo, alivia la sobrecarga del cuidador y retrasa la institucionalización de estos enfermos (ingreso en centros de día o residencias). Se investiga además si estos fármacos pueden ser útiles en otras demencias.
- No farmacológico: muchas personas afectadas, sobre todo en estadíos precoces, pueden beneficiarse de terapias de estimulación cognitiva en las que se potencias las funciones más comprometidas y se desarrollan estrategias de compensación utilizando los recursos cognitivos que todavía están preservados. De esta manera se mantiene un mayor grado de autonomía y funcionalidad. Son recomendables también la terapia ocupacional y el ejercicio físico.
¿Qué es un ictus?
La palabra ictus procede del latín y significa golpe o ataque, y hace referencia a cualquier trastorno de la circulación cerebral, generalmente de comienzo brusco, que puede ser consecuencia de la interrupción de flujo sanguíneo a una parte del cerebro (isquemia) o la rotura de una arteria o vena cerebral (hemorragia).
Popularmente se emplean múltiples nombres para denominarlo: infarto cerebral, trombosis, embolia, derrame cerebral, apoplejía,…, lo que condiciona confusión en cuanto al concepto y la diferenciación entre los diferentes tipos.
El ictus es la primera causa de mortalidad entre las mujeres españolas y la segunda en los varones, según datos del Grupo de Estudio de Enfermedades Cerebrovasculares de la Sociedad Española de Neurología. En Europa mueren 650.000 personas anualmente por esta causa y, de ellos, 40.000 son españoles. Al año se detectan unos 120.000-130.000 casos nuevos. De hecho, cada seis minutos se produce un ictus en España.
El ictus produce más mortalidad y discapacidad que los accidentes y el Alzheimer juntos en España.
¿Cuáles son los tipos de ictus?
a) Isquémicos:
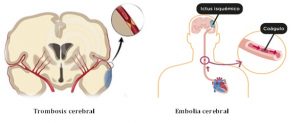
1. Ictus trombótico, aterotrombótico o trombosis cerebral: es aquél en el que el material que obstruye el paso de sangre (trombo) en un vaso se ha formado en el propio vaso.
2. Ictus embólico o embolia cerebral: es aquél en el que trombo está originado por un coágulo de sangre que se ha formado lejos del lugar de la obstrucción, normalmente en el corazón.
3. Ictus hemodinámico: El déficit de aporte sanguíneo se debe a un descenso en la presión sanguínea, por ejemplo, cuando se produce una parada cardíaca o una arritmia grave, pero también puede ser debido a una situación de hipotensión arterial grave y mantenida.
4. Ataque isquémico transitorio: es un tipo de ictus en el que el paciente sufre transitoriamente todos los síntomas con los que cursa un ictus (con frecuencia sólo dura unos minutos), pero éstos desaparecen sin dejar ninguna secuela. También es una emergencia médica.
b) Hemorrágicos:
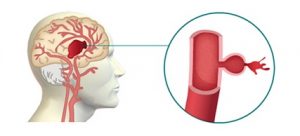
1. Hemorragia intracerebral: se produce cuando una arteria cerebral profunda se rompe y deja salir su contenido sanguíneo, que se esparce entre el tejido cerebral circundante, lo presiona y lo daña.
2. Hemorragia subaracnoidea: es una hemorragia localizada entre la superficie del cerebro y la parte interna del cráneo. Su causa más frecuente es la rotura de un aneurisma arterial (anomalía en la pared de una arteria).
¿Qué síntomas puedo tener?
Los síntomas del ictus son muy variados y dependen del área del cerebro que se vea afectada. La clave para identificar que los síntomas son debidos a un ictus es su inicio brusco.
Entre los principales síntomas que debemos tener en cuenta son:
1. Fuerza: pérdida de fuerza que puede afectar a brazos o piernas, o a la cara (se tuerce la boca)
2. Lenguaje: no pronunciar bien, o lo que decimos es incoherente, confundimos o cambiamos palabras…
3. Sensibilidad: hormigueo o acorchamiento que puede afectar a cara, brazo y/o pierna.
4. Otros: pérdida de visión brusca en un ojo, visión doble, inestabilidad al caminar, dolor de cabeza muy intenso (“el dolor más intenso de mi vida”), etc.
La identificación inmediata de los síntomas del ictus es crucial para salvar la vida y para conseguir las menores secuelas posibles. Se ha demostrado que los pacientes tratados desde el primer momento por neurólogos logran una recuperación casi total o con muy pocas secuelas.

¿Qué hacer y qué no hacer en caso de síntomas sugestivos de ictus?
a) Qué hacer:
1. Avisar a un familiar que me acompañe.
2. Avisar de inmediato al 061 o acudir directamente al hospital de referencia si la ambulancia se va a retrasar.
3. Anotar la hora de inicio de los síntomas.
b) Qué no hacer:
1. Esperar a que se me pase.
2. Tomar Aspirina.
3. No avisar a nadie por no molestar.
4. Avisar al médico de cabecera para que venga a casa a visitarme cuando pueda
NO HAY QUE OLVIDAR QUE ES UNA URGENCIA MÉDICA
¿Qué tratamientos existen para el ictus?
Una de las claves en el control del ictus es la atención inmediata y especializada por un Neurólogo.
El código ictus es un protocolo de actuación prehospitalario o extrahospitalario basado en el reconocimiento de manera precoz de los signos y síntomas de un ictus, con la consiguiente priorización de cuidados y traslado inmediato a un Centro Capacitado de aquéllos pacientes candidatos a beneficiarse de una terapia de reperfusión y de cuidados especiales en una unidad de ictus. Los objetivos del Código Ictus son los siguientes:
- Disminuir el tiempo entre el inicio del ictus y el acceso a un diagnóstico y tratamiento rápido.
Incrementar el número de pacientes con ictus tratados con fibrinolisis. - Reducir la dependencia de los pacientes para realizar las actividades de la vida diaria, la necesidad de cuidados permanentes e incluso reducir la mortalidad.
Cuanto menos tiempo se tarde en llegar a un diagnóstico, más pronto se podrá administrar un tratamiento adecuado con el que se reducirá la cantidad de tejido cerebral que sufra daños irreversibles: TIEMPO ES CEREBRO.
Dependiendo del tipo de ictus y del tiempo de evolución de los síntomas, se aplicarán diferentes tratamientos:
a) En el caso de los ictus isquémicos:
1. Agentes trombolíticos (fibrinolisis): para intentar destruir los coágulos que están produciendo el infarto cerebral y recanalizar el vaso sanguíneo obstruído. Estos fármacos solo se pueden utilizar en las primeras horas del ictus por motivos de seguridad y en unas condiciones muy concretas.
2. Trombectomía mecánica: en determinados casos muy seleccionados, si no es posible administrar la fibrinolisis o ésta no es eficaz, se intenta la extracción del coágulo mediante un dispositivo que es introducido a través de una arteria y con el que se intenta llegar a dicho trombo para retirarlo.
3. Antiagregantes plaquetarios y anticoagulantes: son fármacos que se administran en la mayoría de los ictus isquémicos para prevenir su recurrencia. En función de la causa del ictus y de las características del paciente se determina cuál es el más adecuado para cada persona.
b) En el caso de las hemorragias:
1. Si se producen por alteraciones en la coagulación de la sangre el tratamiento se basa en intentar revertir ese problema si es posible.
2. Si son producidas por malformaciones en los vasos (por ejemplo, un aneurisma) se intenta “desconectarlas” de la circulación mediante cirugía o técnicas endovasculares, para evitar que se puedan volver a romperse y sangrar.
¿Cómo se puede prevenir?
Aunque contamos con medidas terapéuticas para intentar reducir al mínimo la lesión cerebral producida por un ictus, uno de los mejores tratamientos que existen para las enfermedades cerebrovasculares es su adecuada prevención, y ésta comienza por la modificación de los principales factores de riesgo «tratables».
El ictus es más frecuente a partir de los 55 años y su riesgo aumenta con la edad. Se estima que más del 21% de la población mayor de 60 años de nuestro país, casi dos millones de personas, presenta un alto riesgo de sufrir un ictus en los próximos 10 años.
Pero además de la edad, existen otras causas de riesgo como son: la hipertensión arterial, las arritmias cardiacas u otras enfermedades del corazón, la diabetes mellitus, el colesterol elevado, la obesidad, el sedentarismo, fumar y el consumo excesivo de alcohol, entre otros.
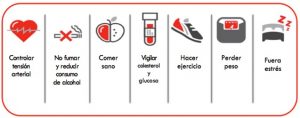
Recomendaciones saludables:
1. EJERCICIO: Ande y haga ejercicio con regularidad: haga ejercicio al menos 3 días a la semana durante al menos 30 minutos. El ejercicio más sencillo , ecológico y con menos riesgo es caminar.
2. DIETA EQUILIBRADA:
- Consumo de frutas, verduras y pescado.
- Alcohol: beba alcohol con moderación, no más de 2 vasos pequeños de vino al día.
- Colesterol: controle su colesterol con una dieta equilibrada, baja en grasas y, como no, con ejercicio!
- Peso: La obesidad se asocia a mayor riesgo de ictus debido entre otras cosas a que la obesidad se asocia a hipertensión arterial, diabetes y cifras más elevadas de colesterol
3. TENSION ARTERIAL: para ello debe controlar el peso, reducir la ingesta de sal en las comidas y… hacer ejercicio!
4. GLUCOSA: controle sus cifras de azúcar en sangre
5. CORAZÓN: cuide su corazón, ya que las enfermedades del corazón son una causa importante de ictus.
6. FUMAR: Nada!
7. FUERA ESTRÉS
Después del ictus
1. Siga las recomendaciones de su Neurólogo.
2. Mantenga hábitos de vida saludables: intentar modificar los factores de riesgo “tratables”, expuestos previamente, es tan importante como la toma de la medicación.
3. Rehabilitación: la rehabilitación busca minimizar los déficits o discapacidades experimentadas por el paciente que ha sufrido un ictus. Se trata de un proceso activo que requiere la colaboración y capacidad de aprendizaje del paciente y de su familia. No solo es importante la rehabilitación física, si no también el apoyo psicológico por parte de la familia y, si fuese necesario, también de un profesional.
La lesión neurológica puede recuperarse por completo, recuperarse solo en parte o no llegar a recuperase nunca, esto va a depender de la gravedad del ictus y de la rehabilitación. Por ello es importante recordar que no siempre es posible conseguir una recuperación completa, y que el principal objetivo es ayudar al paciente a adaptarse.
¿Cómo evoluciona la recuperación?
La mejoría clínica suele ser más evidente en el primer mes y se suele mantener hasta el tercer mes. Entre el tercer y sexto mes dicha mejoría es menor, y con cambios progresivamente menores hasta cumplido el año. En general, se establece que a partir del sexto mes es cuando se produce la estabilización, pero hay que tener en cuenta que las alteraciones del lenguaje y del equilibrio podrían seguir mejorando hasta pasados 2 años.
De forma global se estima que entre los supervivientes del ictus el 44% quedan con una dependencia funcional.
Se debe mantener una vida lo más activa posible, dentro de las limitaciones que existan.
La neurología es la especialidad médica que se encarga de cuidar a los enfermos con epilepsia.
La epilepsia es la principal enfermedad neurológica crónica, ya que afecta al 0.8% de la población en Galicia, lo que supone aproximadamente 18.000 personas. En el mundo se estima que 50 millones de personas padecen epilepsia.
Cuando la mayoría de personas escuchan la palabra “epilepsia” en su mente aparece la escena de una persona que cae al suelo inconsciente, tiene sacudidas incontrolables, babea y pierde el control de la vejiga. Sin embargo, este tipo de crisis, que es la crisis convulsiva generalizada, es sólo un tipo de epilepsia. Hay muchos otros tipos de epilepsia, cada uno acompañado de un conjunto diferente de síntomas.
La epilepsia es un trastorno del cerebro en el cual sus células, que se llaman neuronas, transmiten a veces las señales en una forma anormal. Las neuronas envían señales a otras neuronas y músculos para producir pensamientos, sentimientos y acciones. La epilepsia altera la actividad normal de las neuronas y puede causar sensaciones, emociones y comportamientos extraños, espasmos musculares y pérdida del conocimiento.
La epilepsia puede afectar a cualquier persona, independientemente de sexo, edad, o raza. De hecho algunas personas célebres han padecido epilepsia en la historia, como Julio César, Napoleón Bonaparte, o Agatha Cristie, Isaac Newton o incluso Beethoven.
Los niños y las personas mayores son los que tienen más riesgo de desarrollar la enfermedad. De hecho se calcula que 15 de cada 1.000 personas mayores de 75 años sufren epilepsia.
La falta de conocimientos sobre la enfermedad ocasiona que muchos pacientes con epilepsia sufran discriminación y estigma social. Por ejemplo, en países desarrollados como el nuestro, Alemania, Italia o Estados Unidos únicamente están empleados la mitad de los pacientes con epilepsia en edad laboral, a menudo en trabajos por debajo de su capacidad potencial.
En la actualidad disponemos de numerosos fármacos antiepilépticos que consiguen controlar la enfermedad con un adecuado seguimiento por parte de los neurólogos en hasta un 85% de los casos. Aunque en ocasiones necesitan varios fármacos para poder llevar una calidad de vida plena.
En los pacientes que no se controlan con tratamiento médico es posible en ocasiones realizar una intervención quirúgica o colocar un implante en el nervio vago, similar a un marcapasos cerebral, para que de esta forma puedan estar controlados.
Si padeces epilepsia, o crees que pudieras sufrirla, debes acudir al Neurólogo, que somos los únicos expertos en tu enfermedad.
Los neurólogos estamos comprometidos con el objetivo de mejorar la calidad de vida de nuestros pacientes.
El término trastornos del movimiento engloba un grupo de enfermedades en las cuales predominan las alteraciones en la forma y en la velocidad de los movimientos corporales. La mayoría de estos trastornos están asociados con la disfunción de una estructura cerebral llamada ganglios basales.
Se pueden clasificar en dos grandes grupos:
1.- Trastornos del movimiento hipocinéticos: caracterizados por lentitud del movimiento (bradicinesia) y disminución del movimiento espontáneo (hipocinesia), además de rigidez (aumento del tono muscular por falta de relajación).
En este grupo se engloban:
1.1.- Síndromes parkinsonianos: caracterizados por acinesia y rigidez, bradicinesia (movimientos lentos y de poca amplitud), con o sin temblor. La enfermedad de Parkinson es la causa más frecuetne e importante de parkinsonismo, si bien hay otras causas que se deben tener en cuenta en el diagnóstico diferencial.
Las enfermedades más frecuentes que generan estos síndromes son:
1.1.1.- Enfermedad de Parkinson: es la causa más frecuente de parkinsonismo. Prevalencia de 100-200 casos por cada 100.000 habitantes. La frecuencia aumenta con la edad, sobre todo a partir de los 60 años. Es más frecuente en varones. Se caracteriza por la presencia de: temblor de reposo, bradicinesia, rigidez e inestabilidad postural, pudiendo estar presentes otros síntomas, como el estreñimiento o la hiposmia. El diagnóstico es clínico, apoyado en algunos casos de determinadas pruebas complementarias. A día de hoy no disponemos de cura pero sí de tratamiento para muchos de los síntomas que produce.
1.1.2.- Otras enfermedades que cursan con parkinsonismo:
- Atrofia múltiple de sistemas
- Parálisis supranuclear progresiva
- Enfermedad por cuerpos de Lewy
- Degeneración córticobasal
- Demencia frontotemporal
- Parkinsonismo secundario a fármacos y a toxinas
- Parkinsonismo vascular
- Parkinsonismo postinfeccioso
1.2.- Síndrome de la persona rígida: se engloba dentro de los cuadros clínicos de actividad muscular continua. Se trata de una enfermedad por anticuerpos contra la enzima sintetizadora del ácido gamma-aminobutírico.
1.3.- Síndrome neuroléptico maligno: se trata de una reacción idiosincrásica a determinados fármacos (bloqueadores de los receptores de dopamina) que produce un cuadro de inicio agudo con rigidez intensa, fiebre, taquicardia, sudoración y deterioro del nivel de consciencia.
2. – Trastornos do movemento hipercinéticos: Trastornos del movimiento hipercinéticos: caracterizados por un exceso de movimiento o por movimientos anormales involuntarios (discinesias). Incluyen a las siguientes entidades: temblor, distonía, corea, balismo, atetosis, tics, mioclonías, hiperekplexia (o enfermedad del sobresalto), espasmo hemifacial, esterotipias, acatisia y otros trastornos del movimiento. Muchas veces el diagnóstico diferencial de estas entidades es complejo porque se solapan unas con otras.
Los trastornos más comunes englobados en esta categoría son:
2.1.- Temblor: movimiento oscilatorio rítmico de una parte del cuerpo debido a contracciones rítmicas de músculos agonistas y antagonistas (unos con una determinada función y otros con la contraria). Dentro de este grupo el más frecuente es el temblor esencial. Otras causas de temblor son el temblor fisiológico exagerado, el temblor mentoniano, el temblor relacionado con fármacos, con alteraciones metabólicas (hipertiroidismo), temblor ortostático, temblor cerebeloso, temblor neuropático (entre otros).
2.2.- Distonía: en este trastorno neurológico aparece una actividad muscular mantenida que produce movimientos de torsión repetitivos y posturas anormales. Se produce por la contracción simultánea de músculos agonistas y antagonistas.
2.3.- Corea: consiste en movimientos irregulares, impredecibles, abruptos y breves que cambian de una zona corporal a otra de forma aleatoria. Dentro de este grupo se encuentran la corea de Huntington y la de Sydenham entre otras. Otras formas de corea son la coreoatetosis (movimientos serpenteantes) y el balismo (grado grave de corea con movimientos bruscos y de gran amplitud).
2.4.- Tics: son movimientos bruscos, breves y repetitivos, estereotipados, que varían en intensidad y aparecen de forma irregular. Se pueden suprimir voluntariamente por un período variado de tiempo, pero esta supresión habitualmente se sigue de un período rebote en el que aumenta el número de tics. Una entidad que presenta estos movimientos es el síndrome de Tourette.
2.5.- Mioclonías: son movimientos bruscos, rápidos y muy breves, de amplitud variable, producidos bien por contracciones musculares (mioclonías positivas) o por pérdida brusca del tono muscular (mioclonías negativas y asterixis). Las mioclonías pueden tener múltiples causas, desde alteraciones metabólicas hasta lesiones cerebrales y medulares.
2.6.- Síndrome de piernas inquietas: se caracteriza por la aparición de molestias en las piernas (hormigueos, sensación de intranquilidad, dolor…) que aparecen en situación de reposo, habitualmente vespertinas-nocturnas, que obligan a mover las piernas para conseguir alivio.
2.7.- Otras: Hiperekplexia. Acatisia. Estereotipias.
3- Dentro de los trastornos del movimiento existe un tercer grupo, englobado bajo el nombre de ataxias. La ataxia no es una enfermedad, si no que es un síntoma que puede ser debido a alteraciones en diversas partes del sistema nervioso, más habitualmente en el cerebelo. Se denomina ataxia a la falta de coordinación, que se puede manifestar como una falta de equilibrio al andar (ataxia de la marcha) o una falta de coordinación en lo movimientos de las manos, que se manifiesta con dificultad para alcanzar un objetivo concreto (dismetría).
La ataxia se puede deber a alteraciones en varios de los sistemas que controlan el equilibrio y la coordinación, como son el cerebelo, las aferencias sensitivas o el laberinto y el sistema vestibular.
Así las ataxias se dividen en:
3.1.- Ataxia cerebelosa: por alteración en el cerebelo o en sus conexiones. Cursa con alteración de la marcha en la cual se aumenta la base de sustentación, con pasos irregulares, incoordinación de movimientos y alteración del habla (habla temblorosa, con arrastre de las palabras, llamada disartria).
3.2.- Ataxia sensorial: por afectación de las vías sensitivas que informan sobre la posición de las articulaciones. Puede afectarse por un problema en los nervios periféricos, por problemas en la parte posterior de la médula espinal o bien por alteración de las conexiones entre el cerebelo y la médula. Habitualmente cursa con trastorno del equilibrio que empeora al cerrar los ojos.
3.3.- Ataxia laberíntica: por afectación del oído interno. Hay alteración de la marcha pero no hay alteración del habla ni de la coordinación de las extremidades.
Dolor de cabeza: ¿es frecuente?
En nuestro medio, el dolor de cabeza, también conocido como cefalea, constituye el primer motivo de consulta de los pacientes que requieren asistencia neurológica en las consultas de Neurología.
En un estudio español a más de dos mil entrevistados, un 74% refirió padecer o haber padecido dolores de cabeza a lo largo de su vida. Esta proporción varía entre el 69% de los varones y el 79% de las mujeres, lo que refleja la influencia del sexo en su aparición. Otros estudios elevan la cifra, indicando que hasta un 95% de las personas tendrán dolor de cabeza en algún momento de su vida.
¿Existen distintos tipos de dolor de cabeza?
Sí. Se entiende por cefaleas primarias a las que no son achacables a otro trastorno, mientras que las cefaleas secundarias son apenas una manifestación de otra enfermedad o proceso capaz provocar dolor de cabeza, como pueden son el consumo de alcohol, un proceso febril o padecimientos en la nariz o senos paranasales, entre otros.
Las cefaleas primarias más frecuentes son la tensional (60% del total de dolores de cabeza) y la migraña (también conocida como jaqueca) con o sin aura (15%). Mucho más raras dentro de las cefaleas primarias son las llamadas trigémino-autonómicas, entre las que la principal es la cefalea en racimos o de Horton, que supone el 0,2-0,3% del total de dolores de cabeza.
¿Puede la migraña ser un proceso crónico?
Sí. Entre un 2 y un 3% de pacientes con migraña evoluciona a una situación conocida como migraña crónica, caracterizada por cefalea al menos 15 días al mes, de los que al menos 8 días al mes son de dolor con características de migraña.
Me duele la cabeza: ¿necesito una prueba de imagen?
Probablemente no. Para la mayoría de los pacientes con cefalea son suficientes la historia clínica y la exploración neurológica para llegar al diagnóstico. Solo en determinados casos se necesitarán una o más pruebas complementarias, como pueden ser la tomografía computerizada (también conocida como TAC o escáner) o la resonancia magnética cerebral. El uso rutinario de estas pruebas de imagen se desaconseja en las cefaleas primarias más frecuentes (cefalea tensional y migraña).
¿Es cierto que el dolor de cabeza puede verse agravado por los calmantes?
Sí. La cefalea por abuso de analgesia es una entidad bien conocida y frecuente: afecta al 1-2% de la población general y predomina en mujeres. Consiste en el empeoramiento o cronificación de una cefalea primaria debido al uso excesivo de medicación sintomática empleada para el alivio del dolor inmediato, como son los analgésicos simples (antiinflamatorios no esteroideos, paracetamol, aspirina), opiáceos, fármacos específicos para la migraña (ergóticos o triptanes) o una combinación de ellos. Se considera como un uso excesivo la toma de los analgésicos simples en más de 15 días al mes, o de los otros fármacos en más de 10 días al mes.
Es importante tratar de prevenir esta situación, dado que revertirla puede ser difícil.
El tratamiento de la cefalea por abuso de medicación se basa en la retirada del fármaco responsable, y no es infrecuente que en los días siguientes se agrave el dolor de cabeza y aparezcan náuseas, vómitos, ansiedad e insomnio.
Texto adaptado de la Guía Oficial de Práctica Clínica en Cefaleas de la Sociedad Española de Neurología (2015).
La esclerosis múltiple (EM) es una enfermedad crónica del sistema nervioso central (SNC), potencialmente invalidante y para la que en la actualidad no se dispone de tratamiento curativo. En el mundo la padecen alrededor de 2,8 millones de personas, en Europa cerca de 1.300.000 y en España cerca de 50.000. Galicia es un área con la prevalencia más alta de España y con una incidencia anual algo superior a 8 pacientes por cada 100.000 habitantes, lo que supone que debe haber alrededor de 4.500 enfermos y que se diagnostican unos 19 pacientes nuevos cada mes. Afecta a dos mujeres por cada varón.
Por otra parte, la EM es la enfermedad neurológica no traumática que más discapacidad causa entre los adultos de entre 20 y 30 años, cuando están iniciando sus proyectos vitales y al principio de su vida laboral. Ello da lugar a una disminución de la productividad laboral, de la capacidad de integración social y de la calidad de vida de un grupo de edad especialmente importante desde un punto de vista socioeconómico. Se ha estimado que el coste anual de un paciente con EM es de 26.974 €, lo que hace que esta enfermedad se encuentre entre las 20 que más coste suponen para la sociedad.
¿CÓMO SE PRODUCE?
En la actualidad desconocemos la causa que la produce, si bien el hecho de que su frecuencia sea más del doble mujeres que en varones sugiere una influencia hormonal. Sabemos que no es una enfermedad infecciosa, que no se contagia y que no se hereda. Los sintomas de la enfermedad se producen por una lesión de la mielina – sustancia compuesta por grasas y proteinas que actúa como aislante en las fibras nerviosas y que permite la conducción normal de los impulsos nervioso-. Aunque no sabemos cómo se llega a ello, la idea general es que el sistema inmunológico, que normalmente nos defiende de las infecciones, tiene una reacción equivocada contra alguno de los componentes de la mielina y la daña. En función de en donde se produzca la lesión, aparecerán los diferentes síntomas del paciente. El nombre a la enfermedad se debe a que las cicatrices (esclerosis) que quedan en la zona de la lesión pueden existir en múltiples zonas del cerebro y/o de la médula espinal.
TIPOS DE EM
Aunque recientemente se ha propuesto una nueva clasificación, desde el punto de vista práctico hablamos de formas en brotes (RR: remitente-recurrente) y de formas progresivas (SP: secundaria progresiva y PP:primaria progresiva).
Alrededor del 85% de los casos son formas RR y en ellas el paciente sufre episodios de déficit neurológico que duran al menos 24 horas y que se presentan de forma recurrente con repetición de síntomas previos o con síntomas nuevos. Estos episodios se denominan brotes y tras ellos puede haber una recuperación total o parcial que depende fundamentalmente de la capacidad que tenga el sistema nervioso de reparar la lesión inflamatoria. Actualmente no es posible predecir cuándo ni cómo se producirá un brote de la enfermedad.
En un 15% de casos el enfermo sufre un deterioro neurológico progresivo desde el principio con algunos momentos de empeoramiento brusco con recuperación total o parcial. Son las denominadas formas progresivas primarias (PP) y en su desarrollo predomina el componente degenerativo sobre la inflamación. En estos casos la proporción hombre/mujer es similar y la edad de aparición es más tardía, sobre los 40-45 años.
Entre estas dos formas de la enfermedad está la forma progresiva secundaria (SP), que se presenta en la mitad de los pacientes que han padecido formas RR. Tras muchos años de padecer brotes, el enfermo tiene un empeoramiento neurológico lentamente progresivo, con o sin brotes sobreimpuestos. Esta forma de evolución se ha intentado relacionar con la edad de inicio, con el número de brotes durante los primeros 2 años, con el grado de recuperación tras los brotes, con los síntomas al inicio y con el número de lesiones en resonancia magnética (RM).
SÍNTOMAS
Van a depender de la zona del SNC que se afecte, variando entre pacientes y dentro de un mismo enfermo, y pueden aparecer como un síntoma neurológico único o como una combinación de síntomas neurológicos.
Trastornos visuales. Son los más frecuentes y se caracterizan por visión borrosa o pérdida de visión con o sin dolor ocular. Suele afectarse un solo ojo. Algunos enfermos refieren visión doble, generalmente asociada a otros síntomas neurológicos.
Síntomas motores. Son muy variables, desde una debilidad o pérdida total de fuerza en una extremidad o en una mitad del cuerpo hasta una imposibilidad para la deambulación autónoma. Cuando la enfermedad ha evolucionado puede haber espasticidad, que se describe como rigidez y dificultad para relajar la musculatura, sobre todo de las extremidades inferiores; puede asociarse a espasmos dolorosos que se relacionan con determinadas posturas.
Síntomas sensitivos. Los pacientes refieren sensaciones variables que van desde cosquilleo (parestesias) hasta pérdida de sensibilidad en una mitad del cuerpo (anestesia). Menos veces pueden referir dolor o quemazón en una extremidad, en un área determinada del cuerpo o en una mitad del cuerpo. En ocasiones estos síntomas se ponen de manifiesto con el aumento de temperatura (fenómeno de Uhtoff).
Trastornos de coordinación. Los pacientes refieren dificultad para la realización de movimientos finos y trastornos de la coordinación y/o equilibrio con marcha inestable. Pueden referir visión doble y dificultad para la deglución de líquidos y para articular palabras.
Trastornos genitourinarios. Son frecuentes, sobre todo en las formas más evolucionadas de la EM. Lo más habitual son episodios de urgencia urinaria con micciones frecuentes y sensación de vaciamiento incompleto y menos veces hay dificultad para iniciar la micción y/o retención de orina. El estreñimiento es más frecuente que la incontinencia fecal.
Los trastornos de la esfera sexual también son frecuentes. Además de la disminución de la libido, en los varones suelen caracterizarse por impotencia y trastornos de eyaculación y en las mujeres suele ser disminución de sensibilidad y anorgasmia.
Trastornos cognitivos y emocionales. La alteración más frecuente es la disminución en la velocidad de procesamiento de los datos y en las formas muy agresivas y con largo tiempo de evolución puede haber desde alteración de la memoria y de la atención hasta una demencia subcortical. Algunos fármacos que se utilizan el el tratamiento sintomático de la EM pueden influir en su intensidad o en su aparición.
El trastorno emocional más frecuente es la depresión, generalmente reactiva al diagnóstico o a la situación funcional que es consecuencia de la enfermedad, pero en algunos casos se ha relacionado con lesiones en determinadas áreas del cerebro. Pocas veces, si hay lesiones frontales extensas, puede haber síntomas psiquiátricos y trastornos de conducta.
Síntomas paroxísticos. Es la aparición brusca de un síntoma neurológico como dolor, inestabilidad, hormigueos, trastorno del lenguaje, visión doble, etc, que dura unos minutos y desaparece. No tienen relación con un brote y su tratamiento va a depender de su intensidad y frecuencia.
Un síntoma muy frecuente de la enfermedad es la fatiga. Se describe como “una sensación generalizada de falta de energía” y se produce con esfuerzos mínimos sin que se hayan identificado factores desencadenantes conocidos, aunque empeora con el calor.
DIAGNÓSTICO
Se basa en una historia de brotes con signos clínicos en los que se demuestra la afectación de más de un lugar del SNC. Se dice que “el paciente refiere síntomas unilaterales y tiene signos bilaterales”. Con esos datos, los estudios complementarios que se deben realizar son:
- Análisis del LCR: en la EM existen unas proteínas, las bandas oligoclonales de IgG que indican actividad inflamatoria en el SNC
- Potenciales evocados: los más utilizados son los visuales y es característico un aumento asimétrico de la velocidad de conducción de los nervios ópticos. Si la lesión es muy intensa puede apreciarse una alteración de su morfología
- Resonancia magnética. Se aprecian lesiones múltiples en el cerebro y/o en la médula espinal que pueden brillar tras la administración de contraste. La RM medular puede no realizarse siempre, aunque suele ser necesaria en las formas progresivas.
Además, en todos los pacientes se realizan análisis de sangre y análisis microbiológicos y serológicos para descartar otros procesos que pueden simular una EM y en casos muy especiales pueden necesitarse estudios metabólicos y/o genéticos más específicos.
TRATAMIENTO
Hemos de diferenciar el tratamiento del brote, el tratamiento sintomático y el tratamiento modificador de la enfermedad.
1.- Tratamiento del brote.
No siempre es necesario realizarlo dado que aunque puede acortar su duración no ha demostrado que mejore el pronóstico a largo plazo. Los brotes se tratan con megadosis intravenosas de metilprednisolona durante 3 ó 5 días aunque se puede realizar tratamiento por vía oral con las mismas dosis de ese mismo fármaco. Cuando el paciente no mejora o la recuperación es muy parcial pueden realizarse entre 3 y 5 sesiones de plasmaféresis. No se ha demostrado que las inmunoglobulinas mejoren el resultado de los corticoides.
2.- Tratamiento sintomático.
Persiguen la mejoría funcional y de la calidad de vida del enfermo y no difieren de los utilizados en otras patologías.
- La espasticidad se trata con benzodiacepinas, baclofeno o tizanidina oral, solos o combinados y cuando se asocia a calambres dolorosos se pueden utilizar cannabinoides en spray. En casos seleccionados pueden ser de utilidad las inyecciones de toxina botulínica.
- El tratamiento del dolor puede realizarse con analgésicos habituales y los episodios paroxísticos suelen responder a carbamazepina, gabapentina, pregabalina, fenitoina, amitriptilina o combinaciones de estos fármacos.
- En el tratamiento de la urgencia urinaria los más utilizados son la tolterodina, la oxibutinina, la terazosina o la alfuzosina y cuando la respuesta es nula o insuficiente puede asociarse desmopresina o realizar inyecciones intravesicales de toxina botulínica. El cateterismo vesical intermitente y la cirugía son otras posibles alternativas.
- La fatiga puede responder a antidepresivos como paroxetina y derivados pero en ocasiones responde eficazmente a amantadina, modafinilo o metilfenidato.
- El trastorno de la marcha puede mejorar con fampridina en un 50% de los pacientes.
3.- Tratamiento modificador de la enfermedad.
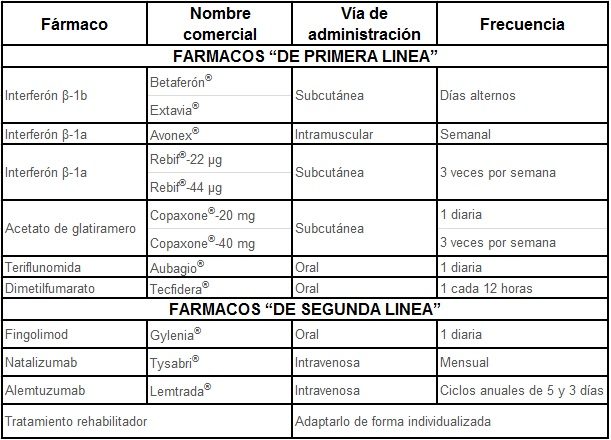
Consigue disminuir la aparición de los brotes, disminuir su intensidad y evitar la progresión de la enfermedad y aunque su eficacia no es absoluta, en los últimos 25 años han demostrado prolongar 10 años la situación funcional de los pacientes cronificando la enfermedad en una situación funcional mejor que cuando no disponíamos de los mismos. Se utilizan en las formas RR de la EM y algunos de ellos pueden administrarse en las formas SP. En la actualidad no hay tratamiento para las formas PP. Estos fármacos tienen indicaciones distintas que dependen tanto de la agresividad y situación funcional del enfermo como del riesgo que supone su administración y en base a estos parámetros se habla de fármacos de primera y de segunda línea. En la tabla adjunta se recogen todos los tratamientos disponibles en nuestra Comunidad.
El tratamiento rehabilitador debe considerarse como una parte imprescindible de la atención al paciente dado que la misma es multidisciplinar y ha de hacerse de forma individualizada adaptándolo a cada caso y modificándolo en función de la evolución del enfermo. Así, en función de cada paciente ha de realizarse fisioterapia, terapia ocupacional, tratamiento neuropsicológico y/o logopedia, sin olvidar que la labor de la enfermería y de la asistencia social también forman parte del equipo que ha de atender a un paciente con un problema neurológico crónico.
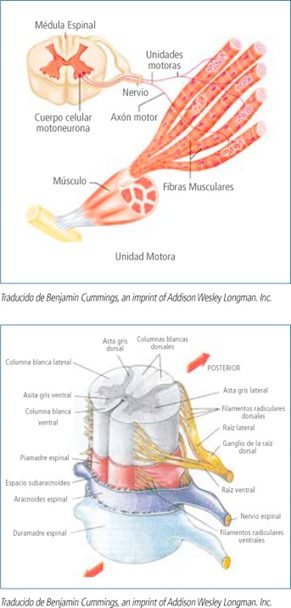
Las enfermedades neuromusculares (ENM) son aquellas que afectan a cualquier componente de la unidad motora (ver imagen), la cual está formada por el músculo, la unión neuromuscular, el nervio periférico y la motoneurona inferior.
Se encuentran dentro del grupo de las denominadas enfermedades raras. En cifras globales, existen más de 50.000 afectados por ENM en toda España.
La mayoría de ellas son enfermedades de origen genético, si bien también pueden ser secundarias a causas adquiridas, como son las enfermedades autoinmunes, inflamatorias o tóxicas.
Dentro de estas enfermedades, la patología más frecuente es la distrofia miotónica, la cual representa un 28% del total.
Las siguientes más frecuentes son las distrofinopatías y la distrofia facio-escápulo-humeral (DFEH), suponiendo un 22.9% y un 10.7% respectivamente.
La atrofia muscular espinal (AME) representa un 5.1%.
Dentro de las neuropatías (las enfermedades que afectan al nervio), entre las más frecuentes se encuentran: la parálisis facial periférica idiopática o de Bell (nervio facial), el síndrome del túnel del carpo (atrapamiento del nervio mediano) o la polineuropatía diabética. Dentro de las hereditarias, la más frecuente es la enfermedad de Charcot-Marie-Tooth.
¿Qué enfermedades se engloban dentro de las enfermedades neuromusculares?
1. ENFERMEDADES DEL MÚSCULO: MIOPATÍAS Y DISTROFIAS MUSCULARES: afectan predominantemente al músculo estriado y son debidas a un defecto en alguna de las proteínas que forman parte de la fibra muscular.
Las miopatías hereditarias y las distrofias musculares son enfermedades de origen genético que pueden iniciarse ya desde la infancia. Provocan una degeneración muscular lenta que da lugar a una debilidad muscular progresiva, con la consecuente discapacidad. Los síntomas principales son la debilidad muscular, la atrofia muscular y las fasciculaciones.
La mayoría de estas enfermedades no tiene un tratamiento efectivo. Es importante mantener actividad física para evitar o mitigar los síntomas motores.
Dentro de este grupo se incluyen las distrofinopatías, las distrofias musculares congénitas y hereditarias, las miopatías hereditarias y congénitas, las enfermedades musculares inflamatorias y las miopatías metabólicas.
2. ENFERMEDADES DE LA UNIÓN NEUROMUSCULAR
2.1 MIASTENIA GRAVIS: Es una enfermedad crónica, autoinmune, de origen desconocido, que se caracteriza por debilidad y fatiga de los músculos esqueléticos (voluntarios) del cuerpo. El problema se encuentra en la unión neuromuscular, encargada de propagar el estímulo eléctrico desde los nervios a los músculos. Esta disfunción se produce por la existencia de unos anticuerpos que destruyen los receptores de acetilcolina bloqueando la transmisión neuromuscular.
Los rasgos característicos de la miastenia son la debilidad muscular fluctuante y la fatiga fácil, que empeoran tras el ejercicio físico y mejoran, total o parcialmente, tras el descanso. La miastenia no produce dolor ni muscular ni articular.
En función de la distribución de los músculos afectados, la MG se clasifica en:
- Ocular: si tras dos años de evolución de la enfermedad la sintomatología solo afecta a los ojos, esto nos puede orientar hacia una forma exclusivamente ocular. Los síntomas típicos son la caída de uno o ambos párpados (ptosis) y la visión nublada o doble.
- Generalizada: si los síntomas aparecen en extremidades (debilidad en las extremidades, alteración de la marcha, dificultad para subir escaleras, dificultad para respirar).
- Bulbar: si afecta a los músculos de la lengua, paladar, masticación y deglución. Da lugar a trastornos del habla (disartria o voz «gangosa»), dificultad respiratoria y dificultad para tragar.
El tratamiento se realiza con diversos fármacos: anticolinesterásicos, inmunosupresores, inmunoglobulinas intravenosas, timectomía y plasmaféresis.
De vital importancia en esta enfermedad es saber que hay determinados fármacos que pueden aumentar la debilidad en algunos pacientes, como son determinados antibióticos (aminoglucósidos, macrólidos, fluoroquinolonas), antihipertensivos y antiarrítmicos y tratamientos hipolipemianes.
2.2- SÍNDROME DE EATON LAMBERT: enfermedad debida a anticuerpos contra componentes de la membrana presináptica de la unión neuromuscular. En un porcentaje elevado de las ocasiones se trata de una enfermedad paraneoplásica, por lo que se debe descartar la presencia de un cáncer oculto.
2.3- SÍNDROMES MIASTÉNICOS CONGÉNITOS: Se trata de enfermedades genéticamente determinadas, que aparecen desde el nacimiento. Se caracterizan por la presencia de fatiga anormal debida a una debilidad muscular localizada o generalizada.
Algunas formas responden de forma parcial a tratamiento con anticolinesterásicos.
Existe una forma adulta de posible comienzo tardío (el síndrome del canal lento).
3. NEUROPATÍAS
Dentro de las neuropatías periféricas, en función del número de nervios afectados y el tipo de afectación, se pueden clasificar en:
- mononeuropatías (solo se afecta un nervio, por ej: síndrome del túnel carpiano)
- polineuropatías (cuando se afectan varios nervios)
- axonales (lesión y degeneración axonal)
- desmielinizantes (lesión de la vaina de mielina)
- neuronopatía (lesión de las neuronas)
La clasificación se basa en el síndrome clínico, los hallazgos patológicos y la etiología. Hay diversas clasificaciones, pero una de las más sencillas y empleadas es dividirlas en agudas y crónicas, simétrica y asimétrica, axonales y/o desmielinizantes.
Existe una amplia variedad de causas que pueden causar daño a los nervios periféricos: carenciales, diabetes, uremia, alcohol, autoinmunes, infecciosas, inflamatorias, fármacos, hereditarias…
Es importante su valoración y diagnóstico precoz, ya que algunos casos son tratables.
4. ENFERMEDADES DE LAS MOTONEURONAS
4.1: MOTONEURONAS INFERIORES: AMIOTROFIAS ESPINALES: grupo de enfermedades caracterizadas por la pérdida o degeneración de las neuronas del asta anterior de la médula espinal. El mal funcionamiento de estas neuronas hace que el impulso nervioso no pueda transmitirse correctamente y, por tanto, los movimientos y el tono muscular se ven afectados. Inicialmente, están mas afectados los músculos proximales y la debilidad en los miembros inferiores suele ser generalmente mayor que la de los miembros superiores. Existen diferentes tipos de Amiotrofias espinales, todas con patrones de herencia autosómico recesivo.
4.2: MOTONEURONAS SUPERIORES E INFERIORES: ESCLEROSIS LATERAL AMIOTRÓFICA: es una de las enfermedades neuromusculares más comunes en el mundo entero y afecta a personas de todas las razas y etnias. La ELA generalmente aflige a personas entre los 40 y 60 años de edad, pero también la pueden desarrollar personas más jóvenes y más viejas. Los hombres son afectados más a menudo que las mujeres. Se trata de una enfermedad neurológica progresiva e invariantemente fatal, que afecta a las neuronas motoras, encargadas de controlar los músculos voluntarios.
Ocasiona debilidad con un rango amplio de discapacidades, afectándose todos los músculos bajo control voluntario. Esto ocasiona pérdida de fuerza y de capacidad para mover el cuerpo y las extremidades. Progresivamente afecta también a la musculatura respiratoria, siendo necesaria la colocación de ventilación artificial si el paciente así lo desea. En la ELA no se afecta la personalidad del paciente, su inteligencia ni su memoria.
Entre el 5 y el 10 por ciento de todos los casos de ELA son heredados. En un 90 a 95 por ciento de todos los casos de ELA, la enfermedad ocurre aparentemente aleatoriamente sin ningún factor de riesgo claramente asociado. Los pacientes no tienen una historia familiar de la enfermedad y no se considera que los miembros de su familia tengan un riesgo mayor de desarrollar ELA.
Hasta la fecha no existe tratamiento curativo. El tratamiento con riluzol ha demostrado un aumento de la supervivencia de varios meses, sobre todo en aquellos pacientes con dificultad para tragar y prolonga el tiempo antes que el paciente necesite soporte ventilatorio. Otros tratamientos están destinados a mejorar la calidad de vida del paciente y a aliviar los síntomas, como son los antidepresivos, laxantes, analgésicos, terapia física, logopedia…
¿Qué síntomas orientan a la existencia de una enfermedad neuromuscular?
- Trastorno de la marcha: las características de la marcha típica de un paciente con enfermedad neuromuscular son el bamboleo, la fatigabilidad, la asimetría, la claudicación y la marcha en punta de pies. Esta marcha es típica de las miopatías y distrofias musculares, aunque también se puede ver en las miastenias y en las atrofias musculares espinales. La fatigabilidad puede indicar un síndrome miasteniforme o a una miopatía mitocondrial.
- Caídas frecuentes: se producen principalmente por debilidad muscular proximal (más cerca al tronco) o distal (más lejos del tronco) de las extremidades inferiores.
- Dificultades para subir y/o bajar escaleras.
- Dolor muscular relacionado o no con el ejercicio.
- Dificultad para ponerse en pie desde el suelo. Este síntoma se conoce como Signo de Gowers. Los pacientes no son capaces de levantarse desde el suelo sin apoyar una o las dos manos en el suelo. Para compensarlo, los pacientes suben apoyándose en sus propias piernas.
- Calambres: son espasmos o contracturas dolorosas. Se observan sobre todo en las enfermedades que afectan al músculo.
- Dificultades de alimentación, atragantamientos. Sobre todo en aquellas entidades que cursan con debilidad de la musculatura bulbar, que permite tragar. Durante meses puede ser la única manifestación del a miastenia gravis.
- Infecciones respiratorias frecuentes y/o prolongadas. Se producen por dificultad para toser y mantener la vía respiratoria limpia. Puede llegar a existir una insuficiencia respiratoria restrictiva.
- Trastornos del sueño: en ocasiones las alteraciones respiratorias pueden manifestarse inicialmente solo durante le sueño, dando lugar a la aparición de despertares nocturnos frecuentes, sensación de ahogo, ortopnea (dificultad respiratoria al estar tumbado), cefalea matinal y cansancio durante el día. Estos síntomas pueden estar indicando una retención de CO2. La entidad que mayor tasa de trastornos de sueño produce es la distrofia miotónica, apareciendo hasta en el 80% de los pacientes.
- Visión doble: por afectación de la musculatura extraocular. Se observa sobre todo en la miastenia gravis, el síndrome de Miller Fisher y en enfermedades mitocondriales.
En la exploración física podemos encontrar:
- Debilidad muscular
- Atrofia muscular, llamada amiotrofia.
- Talla baja o bajo peso
- Cambios en la piel
- Contracturas articulares progresivas
- Escoliosis progresiva
- Hipertrofia muscular. Al contrario de la atrofia muscular, la hipertrofia es un aumento de tamaño de la musculatura esquelética. La musculatura más habitualmente hipertrofiada es la de las pantorrillas, la cual se observa sobre todo en distrofinopatías, al igual que la hipertrofia lingual.
- Contracciones musculares involuntarias: rippling, fasciculaciones y miotonía.
- Hiperlaxitud distal.
- Deformaciones torácicas: pectum carinatum/pectum excavatum. Se produce por compromiso y debilidad de la musculatura torácica.
- Ausencia de reflejos osteotendinosos: en la mayoría de enfermedades neuromusculares, si bien hay entidades que cursan con reflejos vivos, como son determinadas atrofias espinales y enfermedades de motoneuronas.
- Pie cavo: signo clásico de la neuropatía de Charcot-Marie-Tooth.
- Luxaciones de la rótula.
- Temblor fino de manos y temblor de lengua. Por pérdida de neuronas motoras.
- Ptosis palpebral (párpado caído) y oftalmoplejia. Aparecen típicamente en las miastenias, en las miopatías mitocondriales y en algunas miopatías congénitas.
- Paladar ojiva.
- Voz nasal
¿Qué otros órganos se pueden afectar en algunas enfermedades neuromusculares?
El compromiso de otros órganos o tejidos es común en varias de las ENM. Los más importantes a tener en consideración son el compromiso cardíaco, donde se pueden encontrar miocardiopatías dilatadas, restrictivas o alteraciones del ritmo cardíaco con arritmias y fibrilaciones ventriculares, entre otras. Pueden ser causa de muerte súbita y necesitar la implantación de marcapasos o desfibriladores de forma preventiva para evitar muerte súbita.
También se puede afectar la función hepática, con aumento de transaminasas. Suele aparecer en las enfermedades que afectan al músculo, como las glucogenosis y también a las enfermedades mitocondriales.
En determinadas enfermedades pueden existir alteraciones endocrinológicas, como intolerancia a la glucosa, resistencia a la insulina y diabetes mellitus. En algunas miopatías mitocondriales puede existir hipotiroidismo e hipoparatiroidismo.
En algunas distrofias musculares puede existir compromiso cognitivo en grado variable.
Es frecuente también el compromiso ocular y auditivo, que ocurren en pacientes con distrofias musculares, miopatías mitocontdriales.
La constipación crónica frecuente es una manifestación del compromiso gastrointestinal que se produce por
¿Cómo se realiza el diagnóstico de una enfermedad neuromuscular?
Además de la historia clínica y de la exploración física y neurológica, suele ser necesaria la realización de pruebas complementarias para poder llegar a un diagnóstico certero.
Estas pruebas complementarias suelen ser:
- Analítica general con estudio de hormonas tiroideas y enzimas musculares, autoinmunidad, etc.
- Electroneuromiograma: en donde se estudia el funcionamiento de los nervios periféricos, el músculo y la unión entre los dos.
- Punción lumbar: mediante la cual se toman muestras de líquido cefalorraquídeo, cuyo análisis es imprescindible para el diagnóstico de determinadas enfermedades.
- Pruebas de imagen: resonancia magnética, tomografía computerizada (TC). Principalmente se realizan para descartar otras enfermedades que puedan simular una enfermedad neuromuscular o bien para estudio del músculo.
- Biopsia muscular: puede ser necesaria para precisar el tipo de miopatía. Existen cambios musculares característicos en determinadas enfermedades como son las miopatías inflamatorias, la miositis por cuerpos de inclusión, distrofias musculares…
- Estudio genético: cada vez su uso está en aumento permitiendo confirmar y categorizar determinadas enfermedades neuromusculares, en la mayoría de los casos hereditarias.